

Publications & Research
Explore our expertise to help you move forward with confidence, wherever you're headed next.
Featured inRoyal Society of Chemistry
NATURE
American Chemical Society
Angewandte
Chemie
German Chemical Society
Cell
-
Our latest research published in Physical Chemistry Chemical Physics reveals how we've leveraged active machine learning and constant pH coarse-grained molecular dynamics (CpHMD) to discover the first unmodified ultra-short peptides capable of forming stable membrane pores.
Our methodology efficiently navigated the vast 25.6 billion octapeptide sequence space to identify candidates with remarkable membrane-disrupting capabilities. Unlike previous approaches that rely on existing PFP databases, our algorithm learned iteratively through simulation feedback without prior assumptions.
The key innovation lies in our implementation of constant pH molecular dynamics simulations, which accurately capture the critical protonation state changes as peptides transition from aqueous environments into lipid bilayers. This pH-responsive modeling proved essential for reliable predictions, as standard simulations without pH considerations produced false positives.
All 13 lab-tested peptides behaved exactly as our simulations predicted:
11 peptides demonstrated the predicted membrane-disrupting ability
1 peptide formed highly stable, discrete pores with measurable conductance
2 negative controls showed no membrane disruption, confirming prediction specificity
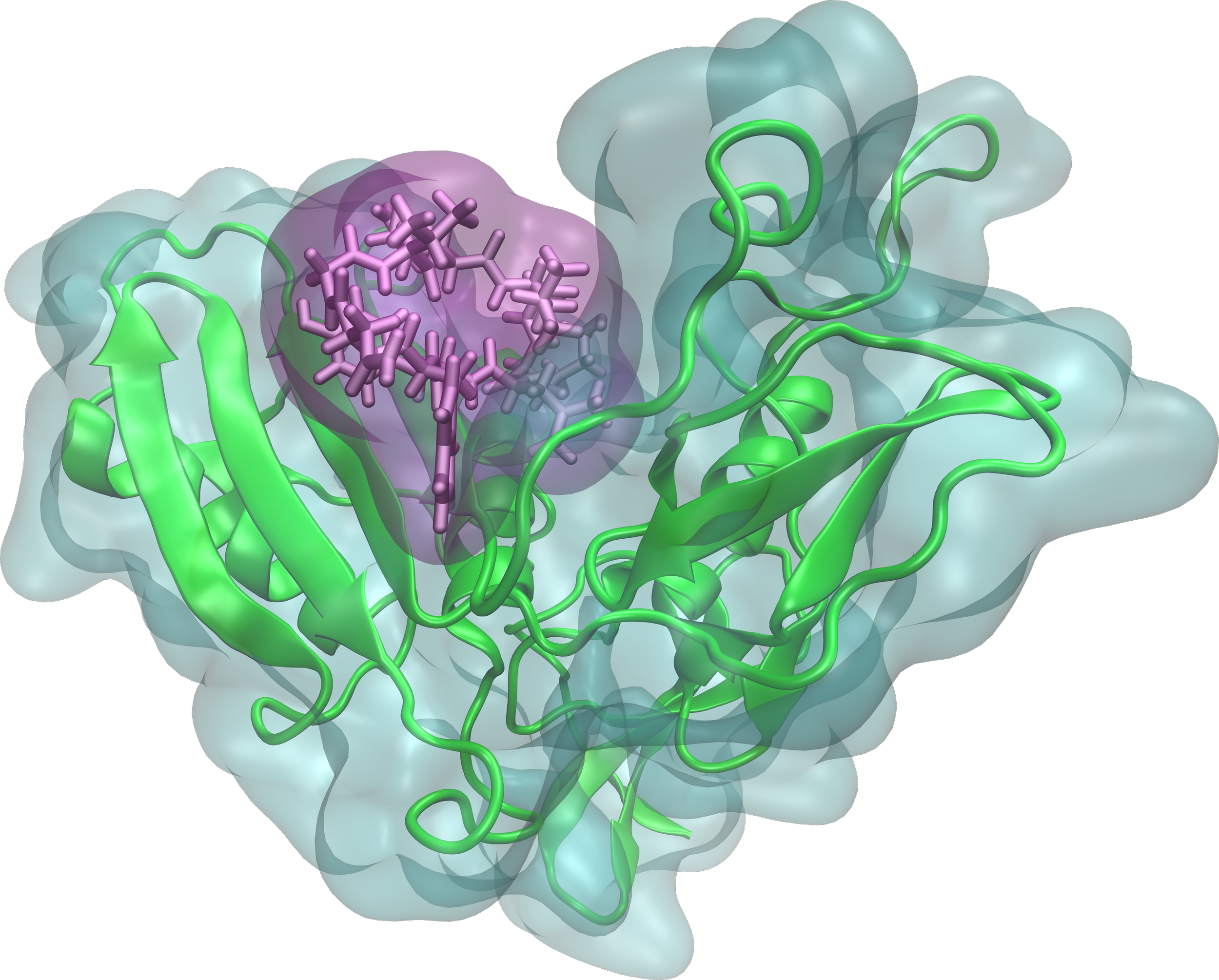
Most significantly, we identified FFMSIRFF as the first ultra-short (8-residue) unmodified gene-encoded peptide capable of forming stable membrane channels, validated through planar lipid bilayer recordings that showed discrete conductance steps matching our computational models.
-
In collaboration with the Wuhan Union Hospital and Huazhong University of Science and Technology we have pioneered a groundbreaking method to repair damaged collagen (the body’s most abundant structural protein) using a custom designed modified ultra-short peptide.
This peptide amphiphile works like a molecular scaffold, guiding broken collagen fragments back into their natural triple-helix form. When combined with nanofat (a stem-cell-rich extract from fat tissue), it creates a robust, bioactive gel that not only restores structure but actively promotes healing.
This material can be 3D printed into precision scaffolds that mimic the body’s own extracellular matrix, supporting cell growth, blood vessel formation, and natural tissue regeneration.
In preclinical models, it out performed traditional fillers by achieving lasting volume restoration, seamless tissue integration and coordinated regrowth of fat and blood vessels bringing us closer to truly regenerative, scar-free repair of soft tissue defects.
-
pH fundamentally influences molecular behavior across biotechnology, biomaterials, and pharmaceutical applications. Our work with constant pH molecular dynamics simulations reveals how pH drives critical biological functions that static models fail to capture.
In pharmaceutical development, drug bioavailability, binding affinities, and degradation pathways all depend on pH-mediated charge states. Targeted delivery systems can leverage the acidic tumor microenvironment for selective drug release, but requires accurate pH modeling to design effectively.
Biotechnology processes benefit similarly from constant pH simulations through optimised enzyme activity, improved protein formulation stability, and enhanced biomaterial stability. The ability to model protonation state changes in complex environments leads to more efficient and precise bioprocesses.
By implementing advanced constant pH methodologies, we bridge computational prediction and experimental reality, enabling smarter materials and more effective therapeutics that respond intelligently to their biological environments.
-
Our clients use the ModelMole platform to create peptide‑based radiotheranostics for overexpressed cancer targets.
These peptides bind to cancer in the body and deliver the radioactive payload directly to the cancer site, maximizing tumor damage while minimizing radiation exposure to healthy tissues.
This approach offers a more effective therapy, and a more economical manufacturing process compared to antibody-based therapies, ultimately leading to improved patient outcomes and increased access to precision radiotherapy.
-
Effective drug delivery systems are critical for improving therapeutic outcomes while minimizing side effects. A key challenge lies in combining both hydrophobic and hydrophilic molecules as conventional emulsions typically accommodate only one type of compound. Additionally, achieving selective drug release at disease sites - such as tumors with their acidic microenvironment - remains an ongoing pursuit in pharmaceutical science.
This study introduces an innovative approach using metal-binding peptides to create switchable emulsions that address these limitations. The system leverages a short peptide sequence (WHHW, Trp-His-His-Trp) that undergoes a structural transformation when bound to metal ions like Zn²⁺ or Cu²⁺. This change enables the peptide to stabilize oil-in-water emulsions capable of co-encapsulating diverse therapeutic agents.
This drug delivery system simultaneously carries hydrophobic drugs (paclitaxel) in the oil phase and hydrophilic compounds (metal ions with the potential to include other biomolecules) at the droplet interface
The histidine-metal coordination breaks down at acidic pH (6.2–6.9), triggering selective drug release in tumor microenvironments.
Emulsion droplets can be functionalised with proteins, antibodies, or imaging agents via histidine tags bound to surface metal ions.
-
This article explores how computation, experiments, and machine learning are revolutionising the design of peptide-based supramolecular materials - a rapidly growing field in materials science and biomedicine. Peptides offer immense potential for creating complex, functional materials due to their ability to self-assemble through non-covalent interactions.
Traditionally, peptide design relied on lab-first, computation second, where researchers would synthesize and test peptides empirically, later using computation to rationalize observations.
However, the field is rapidly shifting toward digital discovery, where computational tools now lead the way in uncovering new materials:
Traditional Approach: Lab-first experimentation, followed by computational analysis to explain observations.
Shift in Discovery: Computational methods and machine learning now lead the process, predicting self-assembly behaviors and identifying promising sequences before lab synthesis.
New Workflow: AI & Computational Chemistry generates hypotheses, while experiments validate and refine predictions - accelerating innovation in peptide-based materials.
M. Ramakrishnan, A.van Teijlingen, et al., Angew. Chem. Int. Ed. 2023
https://onlinelibrary.wiley.com/doi/full/10.1002/ange.202218067
-
Transition state modelling is a longstanding challenge in computational chemistry, often requiring expensive, iterative recalculations and chemical intuition to identify high-energy structures. In this work, we present a faster, more efficient approach by combining umbrella sampling with a machine learning potential (ANI-2x). This allows us to explore the free energy surface and conformational space around transition states with greater efficiency, reducing the computational burden compared to traditional DFT methods.
Why Transition State Modelling is Challenging
Transition states are short-lived, high-energy structures that dictate reaction mechanisms.
Traditional methods like DFT are accurate but computationally expensive, limiting their use in large-scale or long-timescale simulations.
Enhanced sampling techniques (e.g., umbrella sampling) can provide deeper insights but often face computational bottlenecks.
We used ANI-2x, a neural network potential trained on quantum data, to drive umbrella sampling simulations.
This allowed us to rapidly explore reaction pathways while maintaining reasonable accuracy, bridging the gap between speed and precision.
R. J. Urquhart, A. van Teijlingen and T. Tuttle, Chem. Commun., 2025
https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc02090e
